蛋白质折叠中间体3D结构数据的系统性评估
论文信息
| 字段 | 内容 |
|---|---|
| 标题 | Unavailability of experimental 3D structural data on protein folding dynamics and necessity for a new generation of structure prediction methods in this context |
| 作者 | Aydin Wells, Khalique Newaz, Jennifer Morones, Jianlin Cheng, Tijana Milenković |
| 机构 | University of Notre Dame, University of Hamburg, University of Missouri |
| 论文地址 | arXiv:2507.08188v2 / Bioinformatics, DOI: 10.1093/bioinformatics/btag020 |
| 代码地址 | https://github.com/Aywells/3Dpfi 或 https://www3.nd.edu/~cone/3Dpfi |
| 发表时间 | 2026年4月(arXiv v2)/ 已发表于Bioinformatics |
一句话概要
蛋白质折叠是动态过程,但实验3D结构数据几乎全部集中于天然状态,折叠中间体的空间结构数据极度匮乏。本文通过系统文献搜索,仅找到2项后翻译折叠和4项共翻译折叠研究提供中间体3D结构数据,每种研究仅涵盖1个蛋白质。评估表明,设计用于预测天然结构的AlphaFold2在预测共翻译中间体时表现不佳(11个非天然中间体中9个TM-score低于0.5),与简单的“代理中间体”基线性能相当。论文揭示了现有结构预测方法在蛋白质折叠动态建模中的根本局限,呼吁发展新一代中间体专用预测方法。
背景与研究动机
蛋白质折叠是其氨基酸序列逐步形成三维空间结构的过程,折叠中间体是指折叠路径中处于天然状态之前的各种构象。分析中间体对于理解折叠动力学、蛋白质功能以及错误折叠相关疾病(如神经退行性疾病)至关重要。
目前存在两类不同的折叠视角:后翻译折叠(post-translational folding),即蛋白质全长序列在溶液中发生的构象变化;共翻译折叠(co-translational folding),即蛋白质在核糖体翻译过程中逐步延长时发生的构象变化。论文引用了一项近期综述(Duran-Romaña et al., 2025)中的关键发现:大肠杆菌中约三分之一的蛋白质组至少有一个结构域是在共翻译过程中折叠的,在真核生物中这一比例可能更高,因为翻译速率更慢。
核心问题在于: 现有的实验技术——NMR波谱学、冷冻电镜和X射线晶体学——虽然贡献了大量天然状态3D结构数据(PDB中超过20万条结构),但几乎无法有效捕获折叠中间体。原因在于:NMR和冷冻电镜的时间分辨率不足以捕捉微秒至秒级快速波动的构象变化序列;X射线晶体学要求蛋白质形成晶体,而中间体固有的不稳定性和动态特性使其难以结晶。
为什么这个问题值得关注: 虽然动力学和热力学数据(如折叠速率常数、熔融温度、自由能)在一定程度上存在,例如PFDB包含141个蛋白质的折叠速率数据,ProThermDB包含31,580个蛋白质的热力学数据,但这些数据提供的是每个蛋白质的单一量值,而非沿折叠路径的每个中间体对应的量值。它们缺乏揭示蛋白质折叠过程原子级相互作用和结构转变所需的3D空间分辨率。因此,要真正理解折叠动力学,必须获得折叠中间体的3D结构。
研究的核心问题: 既然实验数据匮乏,能否利用已成功预测天然结构的计算方法(如AlphaFold2)来填补这一空白?本文正是围绕这一问题展开系统性评估。
现有方法的瓶颈
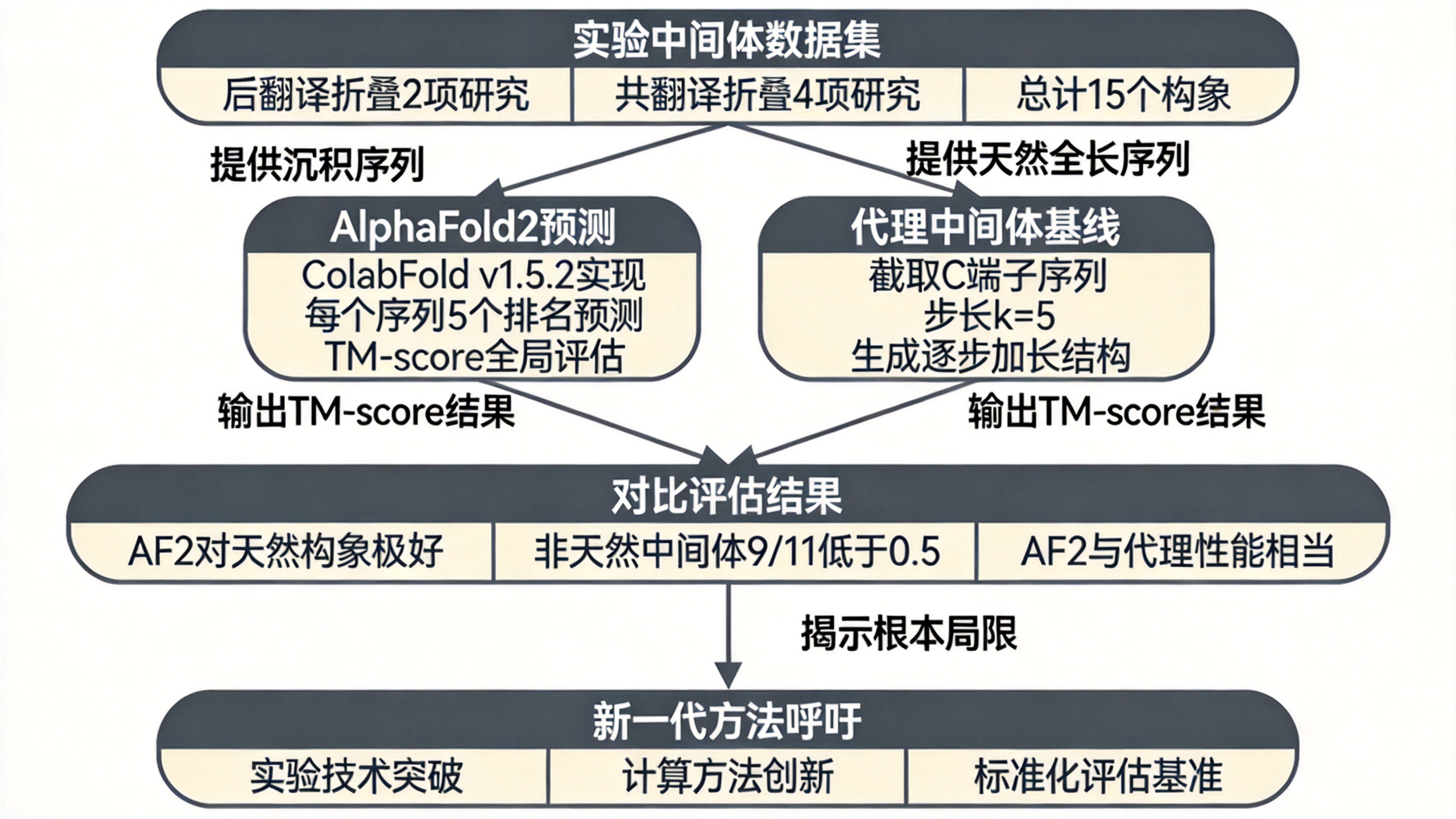
瓶颈一:中间体3D结构实验数据的极端匮乏。 论文的系统文献搜索结果显示:后翻译折叠中间体仅有2项研究提供实验3D结构数据(分别针对Fyn SH3结构域和核糖核酸酶H),每项研究只报告了2个中间体;共翻译折叠中间体仅有4项研究(Agirrezabala et al., 2022; Hanazono et al., 2018; Hanazono et al., 2016; Cabrita et al., 2016),每项研究只涉及1个蛋白质,提供2-4个中间体。所有研究的蛋白质序列长度大多在100个氨基酸以内。这与PDB中超过20万条天然结构数据形成鲜明对比。
瓶颈二:现有天然结构预测方法无法泛化到中间体。 Outeiral et al.(2022)已对后翻译折叠情景进行了评估:他们测试了AlphaFold2、RoseTTAFold、trRosetta等8种方法,结论是这些方法无法有效预测后翻译中间体。在区分二态折叠与多态折叠蛋白质的任务中,所有方法的AUROC得分仅0.56-0.675,低于仅基于链长的简单基线(AUROC=0.739)。在预测折叠速率常数的任务中,链长同样优于所有结构预测方法。
瓶颈三:共翻译折叠中间体的预测研究几乎空白。 论文指出,在本研究投稿之前,未发现任何专门针对共翻译中间体3D结构预测的计算方法。后续有两个研究在投稿后发表(Wang et al., 2025; Tao et al., 2025),分别使用蒙特卡罗方法和分子动力学模拟,但均未通过直接比较预测与实验中间体的3D结构来进行评估。
瓶颈四:天然结构方法用于折叠路径预测时缺乏物理合理性。 这些方法的设计目标本身就是预测最终稳定的天然结构,并未融入折叠动力学生物物理特征(如核糖体几何约束、翻译速率、分子伴侣作用等)。因此,即使通过多次采样生成构象,也难以捕获真正的折叠路径。
核心洞察与贡献
论文的核心洞察在于认识到:现有结构预测方法(包括AlphaFold2)从根本上不是设计来预测折叠中间体的,其对天然构象的精度越高,反而越可能偏离非天然中间体的真实结构。 这一洞察来源于对天然结构预测方法训练范式的理解——它们基于天然构象进行训练和优化,而非基于折叠路径中的瞬态构象。
基于这一洞察,论文提出了系统性的验证框架:
- 贡献1: 首次系统梳理了实验获得的折叠中间体3D结构数据,建立了集中化资源。搜索涵盖后翻译和共翻译两类中间体,明确指出了数据极度匮乏的现状。
- 贡献2: 首次评估了AlphaFold2在预测共翻译中间体3D结构上的表现,通过与实验数据的直接结构比较(TM-score)提供了定量证据。
- 贡献3: 提出了“代理中间体”(proxy intermediates)作为基线方法——从天然结构中截取逐步加长的子结构来模拟共翻译过程,并评估其与AlphaFold2预测的对比表现。
- 贡献4: 揭示了AlphaFold2预测中间体与“代理中间体”在匹配实验数据上表现相当,进一步佐证了天然结构预测方法在折叠动态建模中的根本局限。
- 贡献5: 呼吁发展专门针对折叠中间体的新一代实验技术和计算方法,并提出了评估框架的建议。
方法详解
论文的研究方法可分为四个核心部分。
第一部分:实验中间体数据的文献搜索与整理。 论文通过系统文献搜索,识别出所有明确在PDB中提交了折叠中间体3D结构的研究。对于后翻译折叠,识别出2项研究(Neudecker et al., 2012; Zhou et al., 2008),每项包含2个中间体(1个pre-native + 1个native)。对于共翻译折叠,识别出4项研究,共11个不同的中间体,其中3个中间体报告了多个构象(图3中的Agirrezabala研究:3个构象的中间体1和2个构象的中间体2;Cabrita研究:3个构象的中间体2),总计17个构象。
第二部分:数据质量控制与清洗。 论文识别出两个存在严重数据缺失的构象并予以排除:PDB ID 7OII(建模序列比沉积序列短约50%)和PDB ID 5B3Y(沉积序列仅比其前一个中间体多2个氨基酸,但建模序列丢失大量氨基酸)。最终保留15个构象对应10个中间体用于后续分析。
第三部分:AlphaFold2预测及评估。 对于每个保留的构象,论文将其沉积序列(不含核糖体或麦芽糖结合蛋白部分)作为输入,使用ColabFold v1.5.2(AlphaFold2实现)进行预测。AlphaFold2为每个序列生成5个排名预测,论文主要报告最高排名预测结果,但也验证了所有5个预测结果定性相似且定量几乎一致(图S4)。评估指标选用TM-score(全局结构相似性度量),将AlphaFold2预测结构与实验确定的对应构象进行比较。TM-score低于0.5表示不同折叠,0.5-1表示相同折叠。论文解释了为何选择全局指标而非局部指标:当TM-score低于0.5时,全局折叠已不同,识别局部偏差的生物学意义有限。
第四部分:“代理中间体”的构建与比较。 从蛋白质天然结构的全长序列中,按照步长k个氨基酸截取C末端子序列。第一个代理中间体包含前k个氨基酸及对应的天然子结构,第二个包含前2k个氨基酸,直至包含全长。论文基于其对蛋白质结构分类任务的效果验证了k=5为最优步长(在72个数据集的80.6%中表现最优或接近最优)。比较时,论文仅选择满足两个条件的蛋白质进行研究:天然结构对应最长序列中间体(确保可生成代理中间体)、每个中间体仅有单一构象(避免AlphaFold2对同一序列的多个构象产生相同预测)。满足条件的是Hanazono et al.(2018)和Hanazono et al.(2016)的研究。
实验与结果
实验一:共翻译中间体结构之间的相似性分析。 论文计算了同蛋白质不同构象/中间体之间的TM-score。关键发现包括:同一中间体的不同构象之间TM-score均低于0.5,表明同一中间体的不同实验构象可能具有不同折叠;同一序列在不同时间点的构象变化较大,往往导致折叠变化(TM-score低于0.5);时间间隔越远的构象变化越大。
实验二:AlphaFold2预测共翻译中间体的评估。 这是论文最具核心价值的原创实验。结果明确显示(图4):
- AlphaFold2对天然构象预测极好(TM-score 0.80-0.96)
- 对非天然中间体预测普遍不佳:在11个非天然中间体构象中,9个TM-score低于0.5(0.14-0.47范围),仅2个略高于0.5
- 例如,Agirrezabala研究中CspA蛋白中间体1的三种构象(7NWW、7OIF、7OIG)的TM-scores分别为0.15、0.14、0.17,几乎接近随机相似性(0.17阈值)
- Cabrita研究中FLN5+FLN6的中间体2a、2b、2c的TM-scores分别为0.47、0.53、0.45,其中2b略高于0.5
实验三:AlphaFold2预测 vs “代理中间体”的对比。 论文比较了两种方法在匹配实验中间体时的表现(表1)。结果发现:AlphaFold2和代理中间体表现相当,在所有非天然中间体上都表现不佳(TM-scores 0.21-0.53)。在某些情况下(Hanazono et al., 2018的中间体2),AlphaFold2得分(0.53)高于代理中间体(0.27),但在另一些情况下(Hanazono et al., 2016的中间体1和2),代理中间体得分(0.43、0.46)高于AlphaFold2(0.28、0.21)。这表明两者都不足以有效预测共翻译中间体。
批判性评估:
实验设计的合理性: 论文选择TM-score作为单一指标是合理的,但两个TM-score的轻微差异(如0.53与0.47)是否具有统计学意义未得到讨论。论文报告了所有5个AlphaFold2预测的结果(图S4),显示排名间变异很小(最大差异0.01-0.12),这增加了结论的可信度。
baseline选择的充分性: “代理中间体”作为基线具有启发性,因为它捕捉了结构预测领域的一个常见做法——从天然结构中提取子结构。但这一基线本身非常简单,未能超越它并不能完全否定AlphaFold2的价值,而更多地表明该任务本身对现有方法构成了根本性挑战。
结论对claim的支撑: 论文的核心claim(现有天然结构方法不能预测非天然中间体)得到了充分实验支撑。特别是对于共翻译折叠这一此前未被评估的场景,论文填补了空白。但论文指出的局限是样本量极小(仅4个蛋白质),结论的泛化能力有待更多实验数据验证。
未验证的设计选择: 论文未尝试使用AlphaFold2的高级采样策略(如AFsample的多次采样、子采样策略)来生成构象多样性,也未评估其他天然结构预测方法(如RoseTTAFold)在共翻译折叠上的表现。论文在讨论部分提出了多种替代方案(如重新训练、高级输入调整),但没有进行实证验证。
优势与局限性
优势:
- 填补空白效应显著: 此前对后翻译折叠中天然结构预测方法失败的研究(Outeiral et al., 2022)尚未延伸到共翻译折叠。本文首次提供了共翻译场景下的定量证据,形成了跨折叠类型的统一结论。
- 数据资源化贡献明确: 论文不仅进行了搜索,还对找到的数据进行了质量控制、标准化处理(TM-score计算中的LCCS处理)、以及集中化存储(GitHub仓库)。这为后续研究提供了可直接使用的数据集。
- 批判性视角全面: 论文不仅指出了问题,还系统梳理了可能的解决方案(核糖体几何建模、密码子使用模式整合、分子动力学模拟),并评估了每种方案的优缺点和局限性。
- 可复现性强: 所有实验数据和软件均已公开,代码仓库可用,TM-score计算的标准流程(包括对LCCS的处理)描述详细。
局限性:
- 样本量极小是最大局限。 共翻译折叠的分析仅基于4个蛋白质的10个中间体15个构象。论文自身也承认了这一局限,但认为即使在这样的样本量下,观察到的AlphaFold2失败模式(非天然构象TM-scores普遍低于0.5)具有较强的一致性信号。
- 评估指标单一。 仅使用了TM-score作为结构相似性度量,虽然论文论证了全局指标在本任务中的适用性(因为在折叠改变时局部偏差的生物学意义有限),但加入互补的局部指标(如lDDT)可能提供更丰富的见解。
- 未评估AlphaFold3。 论文选择AlphaFold2而非AlphaFold3的理由是AlphaFold3主要改进在于蛋白质复合物和生物分子相互作用建模,而非单链蛋白预测。但这一判断缺乏实证比较。
- 对实验数据的处理可能引入偏差。 论文排除PDB ID 7OII和5B3Y基于合理的质量控制标准,但排除后仅剩15个构象,进一步限制了结论的泛化能力。
- “代理中间体”比较部分的样本更小。 因筛选条件严格(天然结构对应最长序列、单构象),仅2个蛋白质参与了对比分析,导致该部分结论的统计效力极低。
未来方向与开放问题
1. 实验端的突破方向。 论文指出,目前已有尝试开发新的实验技术,如19F NMR可用于检测共翻译过程中的稳定结构状态。但更根本的挑战在于:如何提高时间分辨率以捕获微秒级的瞬态构象。这可能需要结合多种实验技术(如时间分辨NMR与冷冻电镜的联合使用)。
2. 计算方法的创新方向。 论文提出了三个主要方向:其一是显式建模核糖体几何约束和翻译矢量特性,如Tao et al.(2025)的分子动力学模拟已初步证明核糖体隧道影响新生链构象;其二是整合密码子使用模式作为翻译速率的代理变量,因为同义密码子使用会影响翻译速度进而影响共翻译折叠;其三是利用大型语言模型或扩散模型生成构象多样性,但目前这些模型(如EigenFold、AlphaFold与流匹配结合的方法)同样主要针对天然构象。
3. 评估框架的标准化问题。 当新的中间体预测方法出现时,如何进行有效验证?论文提出了三种策略:与实验3D结构的直接比较(适用于少量已有数据的情况)、与动力学/热力学数据的间接验证(如Outeiral等人的两态/多态分类方法)、独立湿实验验证(如Wang et al., 2025的截断结构域实验)。建立一个标准化的评估基准将极大推动该领域发展。
4. 计算复杂度与可用性的平衡。 论文特别指出,当前文献中常常忽略运行时间和内存使用的报告,新的方法不仅要准确和高效,还应提供直观的界面、良好的文档和开源实现。
组会预判问答
Q1: 样本量这么小(仅4个共翻译蛋白),结论是否有统计效力?
回答: 论文自身承认这一局限,但认为观察到的模式(非天然中间体9/11构象的TM-score低于0.5)具有足够的一致性信号。可以理解为,如果AlphaFold2能有效预测非天然中间体,在多个不同蛋白质上应该至少有一些成功案例,而事实恰恰相反——在所有4个蛋白质上均出现系统性失败(TM-score接近随机)。但必须指出,这一结论的泛化能力确实有待更多实验数据验证,尤其是更长序列和不同折叠类型的蛋白质。
Q2: 为什么选择AlphaFold2而不是最新的AlphaFold3?
回答: 论文给出的理由是AlphaFold3的主要改进在于蛋白质复合物和生物分子相互作用建模,而非单链蛋白质预测(引自Elofsson, 2025对AlphaFold3在CASP16表现的评估)。但这并非直接实验证明。可以理解为,论文在2026年发表,理论上可以更新到AlphaFold3,但考虑到本研究聚焦于单链中间体构象预测,AlphaFold2可能已足够作为代表性方法。若用AlphaFold3重复实验,结果可能与AlphaFold2高度相似,因为两者的核心训练范式(基于天然结构)相同。
Q3: 代理中间体与AlphaFold2的比较部分样本更小(仅2个蛋白),这部分结论可靠吗?
回答: 这一部分的样本筛选标准非常严格(天然结构对应最长序列、单构象),导致只有两个蛋白(Hanazono et al., 2018的λ阻遏蛋白和Hanazono et al., 2016的WW结构域)满足条件。结果的模式为:一个蛋白中AlphaFold2更好(但TM-score仍偏低),另一个蛋白中代理中间体更好。论文的结论是“两者表现相当”,这其实是一种保守表述。可以理解为,该部分实验的设计目的并非证明哪个方法更好,而是论证“即使是简单的代理中间体方法也与AlphaFold2不相上下”,从而进一步支持核心claim——现有方法不足以预测非天然中间体。但4个数据点确实不足以支撑统计推断。
Q4: 论文提到“假设AlphaFold2的高级采样策略和构象多样性生成工具也无法解决此问题”,这个假设有依据吗?
回答: 论文明确将这一判断标记为假设,而非结论。理论依据是:这些工具(AFsample、EigenFold等)虽然能生成多种构象,但其训练和采样仍然偏向于稳定的天然构象。可以理解为,对天然构象的高精度建模可能使得模型在构象空间中形成了吸引子,导致采样局限于天然构象附近的能量盆地,无法覆盖非天然路径中的构象。论文承认这一假设需要实证验证,并将其列为未来工作。
本报告由立理AI生成,仅供参考,请以原文为准。